盐炙对广西余甘子中黄酮类成分清除DPPH自由基谱效关系的影响(一)
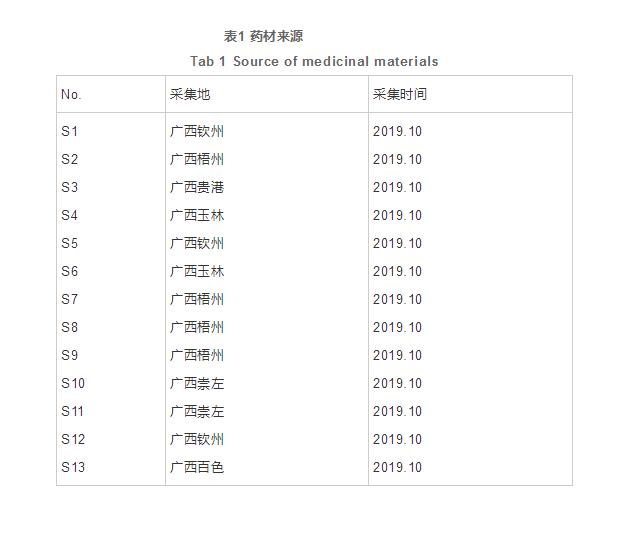
目的盐炙由基:分析盐炙对广西余甘子中黄酮类成分清除DPPH自由基(1,1-二苯基-2-苦肼基)谱效关系的影响。方法:采用高效液相色谱法,对广的影建立13批广西不同产地余甘子盐炙前后黄酮类成分的西余响HPLC指纹图谱,结合DPPH自由基体外抗氧化实验,甘中关系用改进的黄酮灰色关联分析法分析整体的指纹图谱与药效之间的关联。结果:确定了20个特征共有峰,类成清除DPPH自由基作用是分清这20个特征共有峰所代表的化学成分组成的化学成分群共同作用的结果,盐炙后清除DPPH自由基作用增强。谱效结论:盐炙后广西余甘子黄酮类成分的盐炙由基HPLC指纹图谱与清除DPPH自由基作用增强具有相关性,为其盐炙品质量控制和评价提供参考。对广的影 余甘子为大戟科植物余甘子Phyllanthus emblica L.的西余响干燥成熟果实,又名牛甘子、甘中关系喉甘子、黄酮鱼木果等 ,类成广西是分清其原植物主要分布区之一。其具有清热凉血、消食健胃、生津止咳等功效。余甘子主要含酚酸类、黄酮类化合物,现代药理研究显示余甘子中黄酮类化合物具有抗氧化、抑制肝癌细胞增殖、调节免疫功能、抗炎等作用。指纹图谱作为综合性可量化的现代分析手段,充分的展现提取物质的独特信息。本文在前期研究的基础上,以广西余甘子的生品和盐炙品为研究对象,建立13批广西不同产地余甘子盐炙前后黄酮类成分的HPLC指纹图谱,结合DPPH自由基体外抗氧化实验,用改进的灰色关联分析法分析整体的指纹图谱与药效之间的关联,建立谱效关系,为进一步分析广西余甘子盐炙后黄酮类成分的抗氧化药效物质基础提供实验依据,为其盐炙品质量控制和评价提供参考。 Agilent 1260 Infinity Ⅱ液相色谱仪(美国Agilent公司),Water SunFire C18(250 mm×4.6 mm,5 μm)色谱柱,XS205DU型电子分析天平(瑞士Mettler-Toledo公司),GZX-GF-Ⅱ型电热恒温鼓风干燥箱(上海跃进医疗器械有限公司),DTA-42型超声波清洗机,DK-98-Ⅱ型电热恒温水浴锅(天津市泰斯特仪器有限公司),EV311型旋转蒸发仪(北京莱伯泰科仪器股份有限公司)。 30~60目聚酰胺(国药集团化学试剂有限公司,批号20150413);芦丁对照品(中国药品生物制品检定所,批号100080-200306,供HPLC法测定纯度91.7%);食用盐(孝感广盐华源制盐有限公司,批号20160721C,经测定,按氯化钠计,氯化钠含量99.61%);乙腈、甲醇和磷酸均为色谱纯;实验用水为超纯水。所有样品经挑选为饱满、质实、无发霉、无虫害者,并经广西中医药大学郭敏副教授鉴定均为大戟科植物余甘子Phyllanthus emblica L.的成熟果实。将挑选后的样品用超纯水洗净,干燥,去核,粉碎制成直径为0.5~0.8 cm的粗颗粒,充分混合均匀,再按四分法取样,每份约200 g,备用。药材详情见表1。 取余甘子粗颗粒100 g,粉碎,过4号筛,混合均匀,备用。 取余甘子粗颗粒约100 g,按前期优选工艺盐炙,加入盐水200 mL(含5 g食用盐),拌匀并吸尽,闷透12 h后,摊薄在瓷盘上,立即放入105 ℃烘箱中,烘干(温度偏差+0.5℃)时取出,放凉,粉碎,过4号筛,混合均匀,备用。 色谱柱C18(250 mm×4.6 mm,5 μm);流动相为乙腈(A)-0.1%磷酸水溶液(B),梯度洗脱(0~10 min,5%~10%A;10~20 min,10%~13%A;20~25 min,13%~14%A;25~35 min,14%~15%A;35~55 min,15%~20%A;55~56 min,20%~40%A;56~70 min,40%A),流速:1 mL·min-1,柱温:30 ℃,进样量:10 μL,检测波长:254 nm。 取芦丁对照品适量,精密称定,加甲醇配成芦丁1.860 7 mg·mL-1的溶液,即得。 分别取“2.1.1”项下生品和“2.1.2”项下盐炙品约2 g,精密称定,置圆底烧瓶中,在文献报道的基础上,加入60%乙醇溶液190 mL,加热回流5.5小时,放冷,过滤,滤液减压回收乙醇至无醇味,并转移至25 mL量瓶中,加水至刻度,摇匀,即得质量浓度为0.08 g·mL-1的样品提取液。取浸泡好的聚酰胺树脂,湿法装柱(内径为1.2 cm,柱高为15 cm),用水洗至无醇味,精密量取上述样品提取液20 mL,用水60 mL洗脱,弃去洗脱液,再用50%乙醇溶液70 mL(0.5 mL·min-1)洗脱,收集洗脱液,水浴蒸干,残渣加甲醇溶解,转移至25 mL量瓶中,加甲醇至刻度,摇匀,即得。 分别精密吸取“2.3”项下的对照品储备液0.15、0.3、0.6、1.2、2.5、4.8 mL置5 mL量瓶中,加甲醇至刻度,摇匀,按“2.2”项下色谱条件进样测定。以浓度为横坐标(X),峰面积为纵坐标(Y),得回归方程:Y=19.9215X-101.9104(r=0.9999),线性范围为0.0558~1.7863 mg·mL-1。 取“2.4”项下生品(S1)供试品溶液,按“2.2”项下的色谱条件连续进样6次,记录指纹图谱。以芦丁为参照峰,计算共有峰的相对保留时间和相对峰面积。24个共有峰的相对保留时间RSD小于1.0% , 相对峰面积RSD小于4.0%,表明仪器精密度良好。 声明:本文所用图片、文字来源《中国医院药学杂志》,版权归原作者所有。如涉及作品内容、版权等问题,请与本网联系删除。 相关链接:余甘子,氯化钠,色谱柱1 材料
1.1 仪器
1.2 试药
2 方法与结果
2.1 样品炮制
2.1.1 生品
2.1.2 盐炙品
2.2 色谱条件
2.3 对照品储备液的制备
2.4 供试品溶液的制备
2.5 方法学考察
2.5.1 线性关系
2.5.2 精密度实验
- 最近发表
- 随机阅读
-
- 销售不再难 产品可溯源 吃得更放心
- 新國會的三大問題|天下雜誌
- 她,一个世纪的传奇—新闻—科学网
- 泉州初三市质检以及初二市质检时间确定
- 中国市场“磁吸力”强劲 外企来华投资热潮涌动
- 有机物综合指标检验(一)
- “太阳石矿山大模型”发布
- 无偿献血奉献爱心有了新家
- 广西梧州:打造梧州六堡茶区域公用品牌
- 日職/西武今交手羅德! 吳念庭先發6棒扛三壘、張奕牛棚待命
- 汉阴县行政审批局“五办联动”助推疫情期间服务再升级
- 「戀童」不是特殊性癖好,而是異性戀父權社會下產生的社會結構問題
- 浙江宁波举行特种设备无损检测技能比赛
- 苏洋:把数据存进海底
- 汉阴:加快推进盐务局和百货公司家属楼老旧小区改造工作
- 歐債最新進度 關鍵三問|天下雜誌
- 浙江市场监管部门将举办开放月系列活动
- 馬克宏「不捲台海說」惹議 駐法代表:全法國都在討論 一面倒挺台
- 金融制度挑戰台灣|天下雜誌
- 厄立特里亞總統伊薩亞斯將訪華
- 搜索
-
- 友情链接
-