超高效液相色谱法同时测定饮料中的10种食品添加剂(二)
(4)流速的超高测定选择 流速的改变会影响梯度洗脱时间和分离效果。试验考察了0.10~0.60mL/min范围间10种目标化合物的效液相色分离效果。当流速大于0.2mL/min时,谱法品添出峰时间提前,饮料容易造成各目标物之间分离度和峰型都不理想。中的种食当流速小于0.15mL/min时,加剂延长了目标物出峰时间,超高测定降低了工作效率。效液相色所以,谱法品添本试验选用0.15mL/min作为方法最佳流速。饮料 (5)梯度洗脱程序的中的种食选择 试验分析了流动相中甲醇初始比例对分离度的影响。当甲醇比例大20%时,加剂前4种目标化合物出峰较快,超高测定但色谱峰响应值低,效液相色易出现色谱峰重叠现象;当甲醇比例小于20%时,谱法品添各目标物出峰时间延长,并且峰形差,所以选择甲醇的初始比例为20%,0.02mmol/L乙酸铵的初始比例为80%。梯度洗脱程序运行到10min时,甲醇比例再重新回到20%,目的是稳定基线,方便下一个目标物实现更好的分离。 (6)柱温的选择 本试验考察了柱温在20℃~50℃范围间的检测结果,当T<30℃时,各组分分离效果较差1当T>30℃时,柱效降低。因此,选择30℃为最佳检测柱温温度。 在优化的实验条件下,10种食品添加剂在10min内能满足快速分离的需要,且分离度与灵敏度都较好。标准色谱图见图4。 1、标准曲线、检出限的测定 按优化好的试验条件对10种食品添加剂混合标准溶液系列进行色谱分析,以各组分的峰面积为纵坐标,质量浓度为横坐标,并绘制标准曲线。结果表明,在0.050~50.0μg/mL范围内,10种食品添加剂的质量浓度与峰面积呈良好的线性关系,相关系数R2均大于0.999,以3倍信噪比(3S/N)计算方法检出限,方法检出限介于0.0017~0.0034μg/mL之间。标准曲线、相关系数、检出限及线性范围见表2。 2、精密度试验 本试验分别对5.0μg/mL、10.0μg/mL和40.0μg/mL三组浓度的混合标准溶液进行7次平行测定。如表3所示,测定低、中、高三组浓度的相对标准偏差(RSD)范围在0.69%~2.8%之间,表明本试验精密度良好。 取10g饮料样品(精确称量至0.001g)于50mL具塞离心管中,加适量水,超声15min,用水定容至刻度,经高速离心机(4000r/min)离心10min,再用0.22μm微孔滤膜过滤,滤液供UPLC测定。 在样品上进行5.1μg/mL加标回收率试验,每组进行6次平行测定,并与传统液相色谱法进行对照分析,如表4所示,UPLC检测的各组分样品回收率在97.9%~101.1%之间,所有目标物的相对标准偏差均小于5.0%,且两种方法所得到的结果有良好的一致性。 本方法采用超高效液相色谱法测定饮料中10种食品添加剂,在优化后的试验条件下,各组分在10min内可达到完全分离,且具有良好的线性关系,检出限为0.0017~0.0034μg/mL,样品加标回收率为97.9%~101.1%。此方法与传统液相色谱法相比,可大大缩短测定的分析时间,有效提高了检测效率,具有现实指导意义,为应用于食品中添加剂的检测奠定了基础。 声明:本文所用图片、文字来源《中国食品添加剂》,版权归原作者所有。如涉及作品内容、版权等问题,请与本网联系 相关链接:甲醇,乙酸铵,添加剂


三、标准曲线、检出限及精密度测定

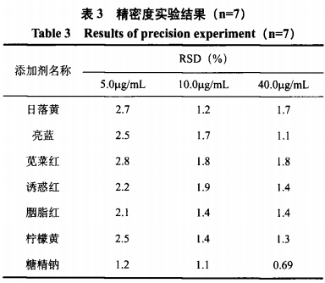

四、样品前处理及样品分析


五、结论
-
上一篇
-
下一篇
- 最近发表
- 随机阅读
-
- “京彩”寒假 志愿暖心
- 磁性纳米粒子辅助加热技术在鱼类解冻中的应用(二)
- 2022年中国漂绿榜
- 前一秒很正常「下一秒覺得自己快死了」!恐慌症發作13症狀 4招緩解
- 汕尾粉签、甘薯盆栽……“百变”陆丰甘薯受追捧
- 银行开户变难为哪般 财经
- 路透:任正非高調回應「5G技術,別人兩三年肯定追不上華為」|天下雜誌
- 天下財經週報:沒有中國電影的金馬獎 是否會更好?|天下雜誌
- 北京丰台:开展夏季水产品安全检查
- 专访港大校长张翔:花10年做一个实验,失败也没什么—新闻—科学网
- 衣服可發光+變色! 香港團隊研發AI光纖布料
- 華為牽手俄羅斯 5G鐵幕隱隱成形|天下雜誌
- 阻止機器人攻擊?馬斯克擬向「X」新用戶收取小額費用
- 杭州亚运筹备按计划推进 场馆已全部面向公众开放
- 以务实举措助力企业做大做强
- 人民幣匯率的「理論值」是多少?|天下雜誌
- 夜场赏花成亮点,武汉“夜樱”搜索量排全国前三
- 以务实举措助力企业做大做强
- 2019年度个体工商户年报报送截止日期临近 尽快报送年报
- 天冷、下雨關節痛!專家教「自救方式」 特別是1運動改善效果佳
- 搜索
-
- 友情链接
-